
英语原文共 4 页,剩余内容已隐藏,支付完成后下载完整资料
对立方卤化物钙钛矿ABX3的电子和光学性质的第一性原理研究
Li Lang, Ji-Hui Yang, Heng-Rui Liu, H.J. Xiang , X.G. Gong
计算物质科学教育部重点实验室(MOE),表面物理国家重点实验室和复旦大学物理系,中国上海200433
摘要:利用第一性原理计算系统地研究了立方相的ABX3型化合物的电子特性。当A或B或X变化时,其性质的化学趋势已被充分研究。ABX3化合物的光学性质也得到研究。我们的计算结果表明,对于精确预测这些卤化物钙钛矿的的带隙,考虑自旋轨道耦合效应是至关重要的。我们预测,CH3NH3SnBr3有一个完美的带隙和较好的光吸收性,是太阳能电池吸收剂中一种有潜力的材料。
1引言
最近,卤化物钙钛矿结构I–IV–VII3族材料(记为ABX3),其潜在的应用引起了广泛的兴趣,例如太阳能电池吸收剂,拓扑绝缘体,甚至超导体。Chung等人通过实验报道了一种新型的全固态型、无机太阳能电池系统,由p型直隙半导体CsSnI3和n型纳米多孔TiO2与染料N719组成,呈现出的转换效率高达10.2%。李等人还报道了一种低成本、可溶液加工的太阳能电池,基于高度结晶的钙钛矿型吸收剂(CH3NH3PbI2Cl)并具有强烈的可见近红外吸收率,,在模拟充分光照下的单一连接装置中具有10.9%的功率转换效率。最近,Burschka等人通过溶液制备得到了基于CH3NH3PbI3/TiO2的光伏电池,拥有前所未有的能量转换效率(约15%)和等于甚至优于当今最好的薄膜光伏器件的高稳定性。所有这些实验表明,这种材料可能是重要的太阳能电池吸收剂。然而,对这种材料的理论研究相当不完整。例如,Borriello等人研究了锡基ABX3化合物的结构和电子性质,Chang等人还研究了铅基ABX3化合物的结构和电子性质。Murtaza等人研究了CsPbX3(X = Cl,Br,I)立方钙钛矿的结构和光电特性。最近,Mosconi等人研究了CH3NH3PbX3和混合卤化物CH3NH3PbI2X(X = Cl,Br,I)的结构和电子性质,并发现了两种不同结构类型的不同性质的混合卤化物钙钛矿的存在。所有这些研究采用局部(或半局部)密度近似,严重低估了带隙。此外,这些研究没有考虑到这些化合物中的重元素的自旋-轨道耦合效应,这个问题直到最近的工作中才被提到。鉴于这些卤化物钙钛矿结构的重要性,对ABX3型化合物进行系统和准确的调查是可取的,对于理解和优化这种太阳能电池吸收材料可能是很重要的。
在本文中,我们利用第一性原理计算系统地研究了一系列ABX3(A = Cs, CH3NH3, NH2CHNH2; B = Sn, Pb;X = Cl, Br, I)型化合物的电子性质,并分析了当A或B或X变化时其化学性能的趋势。我们发现:(i)当A的规模增加时,ABX3的带隙将增加;(ii) 当B从Sn变为Pb时,ABX3的带隙将增加;以及(iii)当x从Cl变化为Br 再到I时,带隙将减小。我们也研究了这些材料的光学特性,并提出CH3NH3SnBr3有一个完美的带隙和较好的光吸收性,是太阳能电池吸收剂中一种有潜力的材料。
2计算方法
我们的计算是在密度泛函理论(DFT)框架中用原子尺度材料模拟程序包(VASP)进行的。500 eV截止能量的赝势增强波投影机(PAW)被用于平面波的基函数。在计算光学性质时采用8 times; 8 times; 8 Monkhorst–Pack k点网格并进一步提高到12times;12times;12。晶格向量和原子的位置根据原子力的指导得到优化,且按标准要求每个原子的计算力小于0.01 eV /Aring;。对于交换相关泛函,Perdew–Burke–Ernzerhof(PBE)的广义梯度近似(GGA)被用来放缓结构参数和计算介电函数,而使用更精确的混合非局域交换相关泛函(HSE)计算电子性质是因为GGA通常低估了这些化合物的带隙。尽管对于Sn(0.43 eV)和Pb(1.32 eV)3P0–3P2的自旋轨道分裂非常大,自旋轨道耦合(SOC)仍包含在所有的计算中。
图1.立方相钙钛矿结构的晶体结构。立方钙钛矿ABX3的原胞是由角相连的X原子的八面体组成,B离子在其八面体中心,A离子在其之间。
表1.计算的ABX3化合物的晶格常数(在Aring;中),(其中A = Cs, CH3NH3, NH2CHNH2; B = Sn, Pb; X = Cl, Br, I)。可用的实验结果显示在括号中。
3晶体结构
通过实验,一些早期的研究报道了在不同温度下ABX3型化合物表现出不同阶段,但在高温阶段,则全部采用立方钙钛矿结构,如图1所示,给出了BX6八面体边共享的三维框架。为了简单起见,在这项工作中我们只考虑该立方结构,以了解这种材料的化学趋势。我们先松弛所有立方ABX3型化合物的结构。所获得的晶格常数如表1所示。我们发现ABX3的晶格常数随着X的大小从Cl, 增加到 Br 以及I而增加。当保持B位置和X位置的原子时,ABX3的晶格常数会随着A位置原子大小而变化。CH3NH3和NH2CHNH2有相似的大小且比CS大,所以A= CH3NH3和NH2CHNH2的化合物晶格常数相接近并且比A=CS的化合物晶格常数大。比较所获得的晶格常数与现有的实验结果,我们可以看到,我们的结果与实验数据符合得比较好。
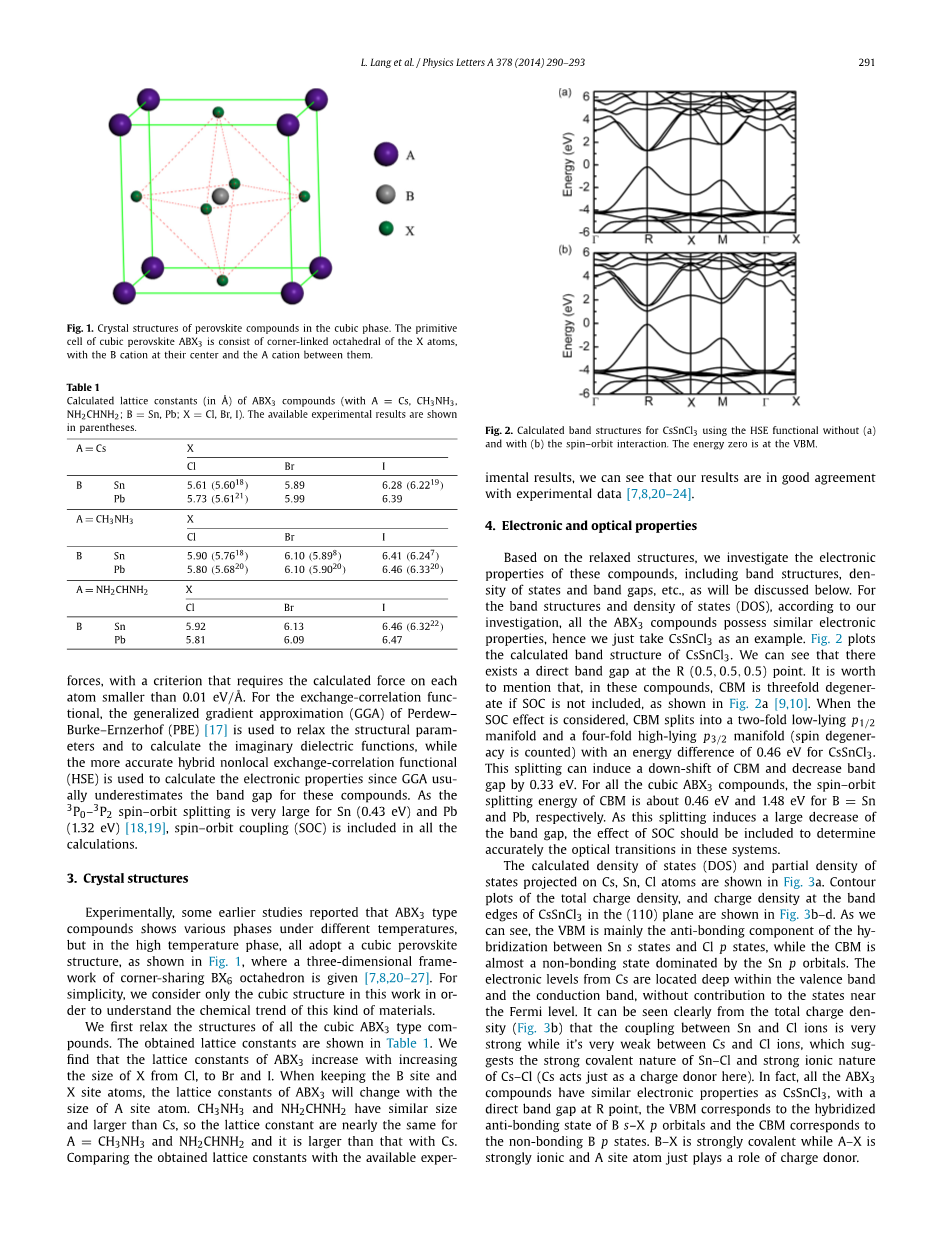
图2.对CsSnCl3使用HSE函数计算的能带结构,(a)图未考虑自旋–轨道相互作用,(b)图考虑了自旋–轨道相互作用。能量零点位于VBM。
4电学和光学性质
基于松弛的结构,我们研究了这些化合物的电子性质,包括能带结构,密度的状态和带隙等,所有这些将在下面讨论。对于能带结构和态密度(DOS),根据我们的研究,所有的ABX3的化合物具有类似的电学性质,因此我们只以CsSnCl3为例。图2 列出了CsSnCl3计算好的能带结构。我们可以看到,在R(0.5,0.5,0.5)点存在一个直接带隙。值得一提的是,在这些化合物中,如果SOC不包括在内,CBM是三重简并的,如图2a所示。当SOC效果考虑,CBM分为双重低能级P1 / 2流形和一四重高能级P3 /2流形(自旋简并数)与cssncl3 0.46 eV的能量差异。当考虑到SOC效应时,CsSnCl3的CBM分裂成一个双重p1/2低能级和一个四重p3/2高能级(自旋简并已算入)且有0.46 eV的能量差异。这种分裂可以引起CBM的下移,并减少0.33eV的带隙。对于所有的立方ABX3的化合物,当 B = Sn和Pb 时,CBM的轨道自旋分裂能量分别为约0.46 eV和1.48 eV。由于这种分裂导致带隙大幅减少,应列入SOC效应来准确判定这些系统中的光学跃迁。如图3a所示,计算出的态密度(DOS)和投射在CS,Sn,Cl原子上的部分态密度。如图3b–d所示为总电荷密度等高线图,和(110)平面图中CsSnCl3禁带边缘的电荷密度图。我们可以看到,VBM主要存在于s态Sn和p态Cl之间的反键结构,而CBM几乎是以非键合态的p轨道Sn为主。Cs的电子能级位于价带和导带的深处,而对费米能级附近的状态没有贡献。从总电荷密度图(图3b)可以很清晰地看出,Sn和Cl离子之间的耦合很强,而Cs和Cl离子之间的耦合非常弱,这表明Sn–Cl的强共价性和Cs–Cl的强离子性(这里Cs表现得就像一个电荷供体)。事实上,就像CsSnCl======一样,所有的ABX3化合物都有相似的电子特性,在R点有一个直接带隙,VBM对应Bs–Xp轨道的杂交反键态,CBM对应B p轨道的无键合态。B–X 是强共价键而A–X 是强离子键, 且 A 位置原子刚好作为电荷供体。
表2.使用HSE函数计算的ABX3化合物的带隙(eV) ( A = Cs, CH3NH3,NH2CHNH2; B = Sn, Pb; X = Cl, Br, I)。括号里得到的结果未考虑SOC效应。
因为GGA通常低估带隙,我们使用混合非局域交换相关泛函(HSE)准确预测带隙。结果列于表2。括号中的数字没有计算SOC效应。我们可以看到,B = Sn 和Pb时,减少的带隙分别为0.35eV和1.13eV。当包含SOC效应时,这表明,在确定这些系统的带隙时,包含SOC效应是很重要的。正如我们所知,带隙在1.1eV和1.5eV之间的半导体作为一个有效的太阳能电池,具有最大的潜力。从这个角度来看,我们可以得出这样的结论:CsSnCl3, CsPbBr3, CH3NH3SnBr3, CH3NH3PbBr3, NH2CHNH2SnBr3 和NH2CHNH2PbBr3由于其带隙在这个范围内,都是潜在的优良的太阳能电池材料。此外,我们还注意到,(i)当A的规模增加时,ABX3的带隙将增加;(ii) 当B从Sn变为Pb时,ABX3的带隙将增加;以及(iii)当X从Cl变化为Br 再到I时,带隙将减小。对于了解和优化这种类型的太阳能电池吸收剂,这些化学趋势是重要的。由上述的能带结构和DOS分析,对于这些趋势的解释如下:(i) 带隙对A离子的依赖性。因为A离子对费米面附近的态没有贡献,它只能间接地通过修改ABX3的晶格常数来影响带隙。因为CH3NH3和NH2CHNH2有相似的大小且比CS大,(CH3NH3)BX3和(NH2CHNH2)BX3的晶格常数十分接近并且大于CsBX3。由于更长的B–X 键,由较大的A原子导致的较大的晶格常数会减少B s–X p的杂化强度。因为VBM是一种杂化的反键态,当耦合强度减小时其能量会降低,因此,这就解释了A原子从 Cs 变为CH3NH3 (NH2CHNH2)时带隙会增加的原因。(ii) 带隙对B离子的依赖性。我们考虑到原子轨道能级。Sn 5s轨道能量比高于Pb 6s,接近X p能级,因此在VBM中Sn s–X p之间的耦合强度大于Pb s–X p。由于VBM是B s 和 X p的反键态, Sn s 与 X p之间更强的耦合使VBM上移并减少了ABX3的带隙。这解释了B 从 Sn 变化为Pb时带隙的增加. (iii) 带隙对X离子的依赖性。由于VBM 包含一些 X p 的特征并且当X离子从Cl变为I时p态能级增加, 从Cl 到I时 VBM 上移并且带隙变得更小。基于这些化学趋势,我们可以使用能带结构工程方法灵活地调整这类材料的电子性质,换言之,通过它们之间的掺杂,就有可能发现或设计性能更好的新型太阳能电池材料。从带隙角度看,由于CsSnCl3, CsPbBr3, CH3NH3SnBr3, CH3NH3PbBr3, NH2CHNH2SnBr3 和NH2CHNH2PbBr3是制造光电池更有潜力的材料,现在我们专注于这些材料,并比较它们的光学性质。在图4中,我们使用PBE方法来计算这些化合物的介电函数,并绘制了虚部(ε(omega;))的图像。由于晶体是各向同性的(A= CH3NH3 或NH2CHNH2对晶体的立方对称性影响不大),ε2的所有对角分量是相同的(εXX =εYY =εZZ),而非对角
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[29014],资料为PDF文档或Word文档,PDF文档可免费转换为Word
课题毕业论文、外文翻译、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。

