
英语原文共 16 页,剩余内容已隐藏,支付完成后下载完整资料
二氧化碳和环己烯氧化物的交替共聚及其与通过N,N配体的锌配合物催化的丙交酯的三聚
Mario Krouml;ger,Cristina Folli,Olaf Walter,and Manfred Douml;ring
摘要:对于CO2和环氧环己烷的交替共聚,一类具有多种氨基亚氨基丙烯酸酯配体的乙酸锌络合物已被合成作为催化剂使用。对于络合物催化反应和结构活性研究揭示,当配体的芳环被不同尺寸的烷基(异丙基与甲基)2,6-取代时,以及当配体骨架包含吸电子基团时达到最高的活性和选择性。这些配合物催化CO2,氧化环己烯和丙交酯的三元共聚可以得到聚(环己撑碳酸酯共聚丙交酯),并且聚合物的组成可通过单体进料调节。
关键词:二氧化碳固定; 共聚;绿色化学; 均相催化; 配体设计; 三聚
正文:由二氧化碳和环氧化合物共聚生产的脂肪族聚碳酸酯因它拥有几个优点得到很多的关注,其中一个主要优点是将CO2 纳入 C链结构,CO2 因为丰富的含量是个诱人和成原料,不同于化石燃料,其资源廉价且不易燃无毒。另一个优点是完美的原子经济性。此外,聚合无需溶剂。最重要的是,生产的脂肪族聚碳酸酯具有一些有趣的性质。例如,其生物降解性允许他们的应用在生物医学领域。但另一方面,CO2反应性低,大多数反应催化剂必须要克服这一问题。
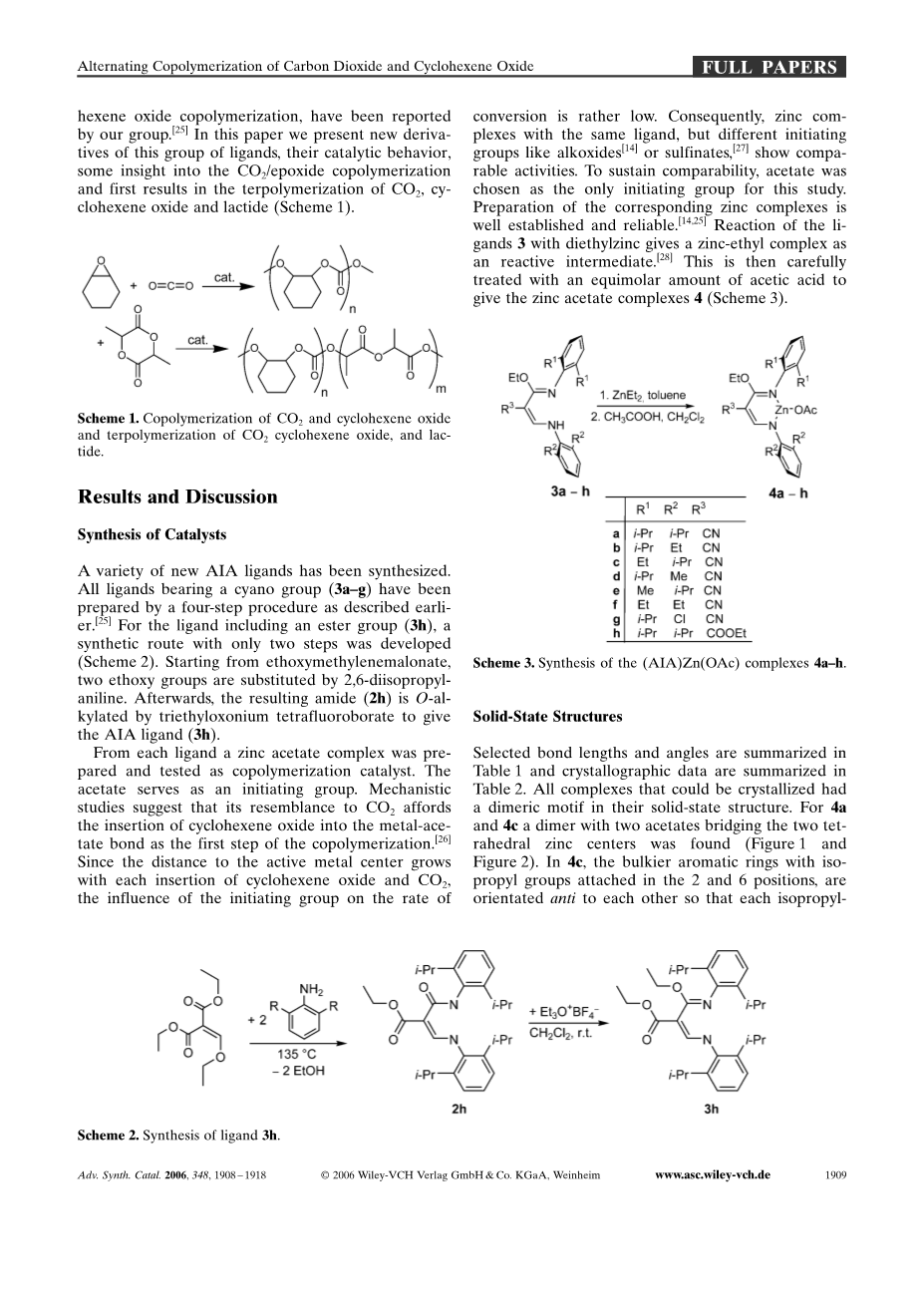
CO2/环氧聚合物共聚是由Inoue在三十多年前发现的,在开始主要使用了非均相催化剂。在过去数十年,均相催化剂的研究导致均相催化活性急剧上升,对于CO2 和环氧丙烷共聚反应,salen 络合物催化剂,特别是与铬和钴形成的催化剂已经成为关注的焦点,然而,到目前为止,CO2 和环氧环己烷交替共聚最高反应活性例子是Coatesrsquo;的b-diketiminato (BDI) 锌配合物 (见下文),因为它与金属有很强的结合、可调节的空间结构和多变的连接方式,他是均相催化剂中重要的一部分。此金属络合物被应用于比如烯烃的低聚和聚合,甲基丙烯酸甲酯的聚合,丙交酯的开环聚合。目前已合成了此配体的四配位的衍生物键合稀有金属得到的刚性金属锌醋酸复合体,作为传统b-diketimines替代物,催化CO2与环氧聚合物的共聚,新aminoimidoacrylate(AIA)配体和他的乙酸锌络合物被我们小组在CO2/环己烯氧化共聚有高的活性。在本文中,我们提出了这系列配体的新的衍生物在CO2/环氧环己烷共聚和CO2,环氧环己烷和丙交酯三元共聚反应初步催化行为。(图一)
图一:CO2和环己烯氧化物的共聚和CO2环氧己烷和丙交酯的三聚。
目前已经合成各种各样的新型AIA配体,通过前面描述的四步合成的带有氰基的配体(3a–g),对于包含酯基(3h)的配体,如今开发出一种2步的合成路线(图二),起始于乙氧基亚甲基丙二酸,2个乙氧基基团被2,6-二异丙基苯胺取代,然后,将所得酰胺(2h)通过三乙基氧鎓四氟硼酸进行O-烷基化,得到AIA配体(3h)。。
图二:合成3h配体
把制得的醋酸锌络合物作为共聚催化剂进行测试,乙酰氧基作为一种引发基团,机理研究表明,其与CO2的相似性使得环氧环己烷插入金属 - 乙酰氧基键,这是共聚的第一步,由于与活性金属中心的距离随着环氧环己烷和二氧化碳的每次插入而增长,起始基团对转换率的影响相当低,因此,锌金属络合物和一样的配体但是不一样的引发基团例如亚磺酸盐和金属醇盐表现出来不同的活性,为了保持可比性,乙酰氧基被选为唯一的引发基团,通过明确的方法制备锌配合物。配体3与二乙基锌的反应得到锌 - 乙基络合物作为反应性中间体。然后通关等摩尔量的乙酸反应以得到醋酸锌络合物(图三)
图三:(AIA)Zn(OAc)络合物4a-h的合成
固态结构:所选的键长度和角度已总结在表格1中,催化剂数据在表格2中,所有可以结晶的络合物在其固态结构中具有二聚体,对于4a和4c研究发现,两者结构都是具有两个乙酰氧基桥接两个四面体的中心形成二聚体,(结构图一和结构图二),在4C中,苯环在2和6号位上取代异丙基,这两个异丙基彼此反向,所以相比较乙基取代有更小的空间位阻。因为空间位阻减小, 4c的Zn-Zn键长(3.963 Aring;, 表一)比4aa (4.161 Aring;)短,在配体主链上可以发现两种结构之间的另一显着差异,在4a中,氨基 - 亚氨基丙烯酸酯骨架的C-C单键和双键与对应的原始单双键长相符(C13-C14:1.368 Aring;, C14-C15: 1.495 Aring;),于此相反的是,4c的配体的主体的C碳碳单键和双键长度显示了一种处在共轭pi;电子体系中的长度特征,单键和双键有着接近的长度,对于C-N键也是接近碳碳单键与双键的长度。
表一:选择的距离(Aring;)和角度(deg)
表二:结晶数据和结构细化细节
结构图一:复合物4a的球棒模型(25%含量)。
结构图二: 复合物4c的球棒模型(25%含量)
其他两种固态结构源于晶体不经意暴露于空气和水,尽管获得偶然,但是揭示了这类锌络合物易与空气和水结合,5d(结构图三)源于对4d 结晶,从结构看,它与醋酸锌络合物的结构类似,但是一个连接锌原子的乙酰氧基被羟基取代。相比4a和4c,其配体上是小体积的甲基取代苯环,同时基团空间上彼此反向。在这种情况下,这两个锌中心都被分别连接一个乙酰氧基和一个羟基,与4c相似,配体的主体的键长度显示了在共轭体系中的特征。与4a和4c的八元环比较,两个锌中心和相连配体组成6元环,因此 Zn-Zn键长明显减小,这种六元环配体结构符合其他关于b-diketiminato锌配体学说,这种六元环比乙酰氧基桥接的八元环或烷氧基桥的四元环有更好的热力学稳定性,因此,这样一种从八元环到六元环的转变因该是将环氧环己烷插入金属与乙酰氧基键之间的驱动力。不幸的是,尝试使乙酸锌络合物与环己烯氧化物反应,没产生适合于X射线分析的晶体,但是相似的结构已经被Moore等得到。
6e的结构更加复杂(结构图四),他有两个不同的部分连接形成无休止的链(结构图五),在一边,它拥有一个与5d同构的部分,两个锌中心被一个乙酰氧基和羟基链接形成一个六元环,同样,配体骨架所有的c-c键显示处于共轭体系的相似的长度特征。同时,苯环被两个相反方向的异丙基取代,但是该结构包含了一个意料外的结构,另一部分结构包含7个锌原子,其中中心锌位于反转中心,整个结构可描述为两个四面体的Zn4单位,其链接通过一个锌原子,每个Zn4中间被放置于一个氧原子,其氧原子与四个锌原子相连接,不在反转中心的三个锌原子是处于一个扭曲的四面体环境,而不是位于中间位置,其中的两个参与3个乙酰氧基链接,只有第三个,是唯一一个参与两个乙酰氧基连接的的。并通过与配体的CN部分相连接完成他的配位层,有趣的是,一些相似的镍配合物的配体有氰基-金属配位但是没有形成预计的N-N螯合物。Zn7单元的中心锌原子处于一个扭曲八面体中,其与两个四面体的中心氧原子再加上四个氧原子配位而不是连接上配体的N原子,整个第二部分通过氰基和两个相应的锌原子连接到两个配体,整个结构形成聚合物带。类似的结构曾被观察过许多次,但是其单位的形成要在锌,醋酸,水的条件下,整体来看,一个这样的结构单元链接7个锌原子,这是潜在的活性催化剂中心。
结构图三:[(3d)Zn(m,h2 -OAc)(m,h2 -OH)Zn(3d)] (5d)的球棒模型
图四:[(m,h2 -OAc)5(m,h4 -O)Zn4(m,h3 -3e)Zn(m,h2 -OAc)(m,h2 -OH)]1 (6e).的球棒模型
图五:[(m,h2 -OAc)5(m,h4 -O)Zn4(m,h3 -3e)Zn(m,h2 -OAc)(m,h2 -OH)]1 (6e)的带状结构图
共聚反应实验:总的来说,所有测试的催化剂都能够催化CO2和氧化环己烯的交替共聚,产物聚环己基碳酸酯(PCHC)的碳核磁图得到全同和间同碳酸酯随机分布,因此聚合物都是无规的, 114℃玻璃化转变温度也与文献描述相符。使用一些最活泼的系统(4d,4e)也一起测试了CO2和环氧丙烷的交替共聚,但是这些反应都只发现了痕量聚碳酸酯的存在。到目前为止,只有一些特别的基于锌配体的b-diketimine催化剂有巨大活性。CO2和环氧环己烷的交替共聚在催化剂荷载用0.1%在CO2压力4MPa不用其他溶剂的条件下进行。优化实验证明了80℃到100.℃是理想的温度区间,在更高温度下,越来越多的环状环己基碳酸盐形成,这是热力学优势的副反应。,在100℃以下实验环己基碳酸酯产生率不超过10%,在更低的温度下,催化剂活性降低,另一方面,因为环氧化合物的活性远远高于CO2,环氧化物均聚生成聚醚的反应,要抑制醚的产生,一个理想的共聚催化剂应该一点也不催化聚醚的产生,大多数提出的催化剂显示了良好的选择性,在90℃下聚醚的形成不会超过12%到14.%(表三),但对称取代的催化剂观察到高聚醚形成,如两个苯环上都是2,6-异丙基(4a,4h)或2,6-乙基(4f)取代的催化剂。总之,这些催化剂活性低,把骨架上的吸电子基团用乙酸乙酯基团替代得到催化活性变小的结果。另一方面,取代模式差异会引起催化活性巨大的差异,当一个环上2,6被异丙基取代和另一个环上2,6位被甲基取代时(4d,4e)催化剂活性最高且最有选择性。催化剂4e最大催化活性TOF超过200h -1,这和b-二酮亚胺锌络合物表现十分接近,带有异丙基和乙基取代基(4b,4c)的不对称催化剂的活性略低,无论是大的环在大的亚胺酸酯一侧或相反,两个芳环化学环境只是略微不同,催化剂活性相近。为了看看芳环上的吸电子取代基是否会增加活性,设定4g,在一个苯环2,6位上引入氯。虽然引进吸电子基团上的配体骨干通常也会增大活性,但是,4g催化剂的活性低于对称不活跃烷基取代的催化剂。氯基产生相当小的影响的原因可以归咎与其基团处于芳环上。配体主体的共轭电子体系是直接与活性金属中心接触,总得来看,催化剂4e有着最高的活性,一般来说,催化剂活性越高产生的聚碳酸酯有更高的分子量(Mnasymp;20000)更窄的分子量分布(PDIasymp;1.2),狭窄的分子量分布显示这是活性聚合,这通过动力学研究证实,所得聚合物的单体(CHO)转化率和链长度之间存在线性关系。假设高分子链从一个活性金属中心开始增长,单体与催化剂的比例是已知的,则可以计算每个阶段单体转化率对应的聚合物长度。有趣的是,这种计算仅符合当在一个活性金属中心上生长两个聚合物链时的实验结果
这与上述的结论(如固态结构)相反,这种结论相当支持双金属增长理论,因此需要进一步的研究来解释这个问题。
表三:催化剂在CO2 /环己烯氧化物共聚中的性能
a:整个使用1000:1的[CHO]:[Zn]比,b:每摩尔锌消耗的CHO摩尔数(每小时TOF),c:通过1 H NMR测定,d:通过GPC在THF中测定,e:如文献[14]中所报道的合成。
三元共聚合实验:第一步实验仅采用外消旋二丙交酯和络合物4e,4e在催化LA形成PLA时显示高活性,然后用催化剂4d和4e分别催化 CO2、CHO和LA三元共聚,大多数反应条件维持与共聚条件类似,只是催化剂浓度上升至0.25%同时反应时间提升至16h。在两种络合物催化的反应里,产生的PCLA含有大量PLA链段(表格四),为了PPC链段含量增加,加入3倍过量的CHO,因为反应条件没有优化,催化剂TOF值仅为中等水平,但是得到高分子量低分布的产物,通常,与S-丙交酯的反应相比,用外消旋丙交酯的反应使催化剂活性变高和得到更高的聚碳酸酯含量。用S-丙交酯的三元共聚反应产生具有较大PLA级分的半结晶共聚物。所得半结晶共聚物熔点为167 ℃,至于上述共聚催化剂4e,4e小体积芳环(2,6-甲基取代)在大的亚胺酸基团同侧,4e是最活泼的催化剂可产生大量的聚碳酸酯链段。尽管之前已经使用非均相催化剂催化与CO2,PO和己内酯的三元聚合,但据我们所知,之前没有进行过CO2,CHO和LA的三元聚合。之前的产物只能通过环状碳酸酯和丙交酯开环聚合得到(结构图四),其缺点是冗杂的环状碳酸酯单体制备。对于文献已知的BDI锌配体催化剂活性的比较实验在相同条件下进行,
表格四:催化剂在CO2 /环己烯氧化物/丙交酯三元聚合中的性能
a:所有三元聚合实验在90℃,4MPa和0.1mol%下进行16小时。 催化剂负载
b:每小时每摩尔锌消耗的CHO摩尔数
c:通过1 H NMR测定,与100%的差为醚含量。
d:通过GPC在THF中测定
e:通过差示扫描量热法(DSC)测定。
结构图四:催化剂(BDI)Zn(OAc)
众所周知这种络合物催化丙交酯的聚合具有选择性,我们发现其在催化三元共聚反应也具有选择性,如前所述,必须使用过量的CHO单体以在
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[139073],资料为PDF文档或Word文档,PDF文档可免费转换为Word
课题毕业论文、外文翻译、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 聚苯胺的制备及其在超级电容器方面的应用文献综述
- 亚苯基主链萨伦铬的制备及催化二氧化碳共聚合的研究文献综述
- 镧合并ß-Ni(OH)2纳米阵列用于稳健的尿素电氧化外文翻译资料
- 基于Lewis碱性吡啶的金属有机荧光探针外文翻译资料
- 软配体在纳米粒子超晶格组装中的作用外文翻译资料
- Janus膜具有可控的不对称配置,用于高效的油乳液分离外文翻译资料
- 具有 Janus 表面的中空纤维膜用于水包油乳液的连续破乳和分离 ——副标题外文翻译资料
- 具有芴基卡多结构的聚苯硫醚具有高透明度、高折射率和低双折射外文翻译资料
- 聚碳酸酯聚合物中芴的聚合度与光热性能的关系外文翻译资料
- 通过狭窄的纳米孔与孤立圆孔连接的阶级多孔膜:一种新颖的在分离中解决权衡效应的解决方案外文翻译资料

