基于蝴蝶形给体—受体发色团的,并具有强固态荧光和大量辐射激子高效近红外有机发光二极管
原文作者 Liang Yao, Shitong Zhang, Rong Wang, Weijun Li, Fangzhong Shen, Bing Yang, and Yuguang Ma
摘要:近红外(NIR)有机发光二极管(OLEDs)的发展受到越来越多的关注。供体—受体(D-A)发色团是近红外OLED中一类重要的近红外材料。然而,基于传统D-A发色团的近红外有机发光二极管的外部量子效率(EQEs)通常低于1%。本文报告的是蝴蝶形的D-A化合物PTZ-BZP。PTZ-BZP薄膜显示出强烈的近红外荧光,其发射峰在700 nm处,对应的量子效率达到16%。值得注意的是,基于PTZ-BZP的近红外OLED的EQE为1.54%,并且观察到低效率的衰减,以及高达48%的辐射激子比,突破了传统荧光OLED 25%的极限。为了了解PTZ-BZP的激发态特性,进行了实验和理论研究。
关键词:发色团;供体—受体体系;近红外荧光;光电材料;有机发光二极管
有机发光二极管(OLEDs)可以发出可见光和白光,在过去的20年里在平板显示器和固态照明领域取得了巨大的成功[1]。由于光学传感、夜视设备和信息安全显示器的迫切需求,将OLEDs的光谱从可见光扩展到深红色和近红外(NIR)已经成为OLEDs领域的新目标[2]。有几种类型的有机或金属复合材料用于近红外OLEDs,包括镧系金属配合物[3],共轭聚合物[4],磷光金属配合物[5],具有给体—受体(D-A)结构的有机p共轭化合物[6]。这些近红外OLEDs的外部量子效率(EQEs)通常低于1%。最近,近红外磷光OLEDs(POLEDs)的EQE取得了重大突破:一种扩展p共轭的铂(II)卟啉的EQE高达9.2%。然而,这些近红外POLEDs的高EQE值通常出现在非常低的电流密度下,并且在高电流密度和高亮度下存在严重的效率衰减,这可能是这种近红外OLEDs中三重态激子的长寿命造成的[7]。
考虑到无金属有机材料的荧光性质(避免长寿命激子的猝灭)和成本优势(不含昂贵的金属),开发无金属有机材料具有重要意义。目前的研究主要集中在具有给体—受体(D-A)结构的p共轭化合物上,因为强的D-A相互作用可以导致更窄的带隙,从而导致深红色或近红外光谱范围内的电子跃迁[8]。然而,这些D-A化合物在光致发光和电致发光(EL)器件中都表现出了意想不到的低效率。低效率的主要原因是内在限制,根据能隙定律预测,由于基态和激发态的振动重叠,发色团的量子效率随着能隙的减小而降低[9]。外,D-A发色团中的HOMO/LUMO重叠通常是有限的,这往往导致较低的辐射跃迁率,最终导致几乎没有发射[10]。体薄膜中,由于这些极性发色团的偶极-偶极相互作用引起的无辐射过程,进一步降低了荧光效率[11]。此,对于高效的近红外OLEDs,D-A发色团必须具有合理的辐射跃迁率,并且在固体薄膜中尽可能保持荧光效率。更重要的是,进一步提升基于D-A发色团的荧光近红外OLEDs (FOLEDs)的EQE需要一种创新的分子方法。辐射激子比率的增加和三重态激子的使用可能具有巨大的潜力,最近报道的一些D-A化合物就证明了这一点[12]。
在此,我们报告了一种蝴蝶形的近红外D-A化合物,PTZ-苯并二唑(方案1),其中吩噻嗪作为电子供体,苯并噻二唑作为电子受体。DFT优化的基态几何结构(B3LYP/6-31G(d, p)方法)揭示吩噻嗪部分具有非平面的“蝴蝶状”结构,C-S-N-C二面角(theta;N)为142° [13]。此外,吩噻嗪和苯并噻二唑基团以145°的扭曲角(theta;D-A)扭曲连接,这是D-A化合物的相对平面排列。通常,由于供体和受体部分的空间分离,电荷转移(CT)激发态的电子跃迁被部分禁止。在我们PTZ-BZT的例子中,吩噻嗪和苯并噻二唑基团的近平面排列(theta;D-A)可以引起这些基团之间的轨道重叠,从而进一步提高辐射跃迁率和荧光效率。
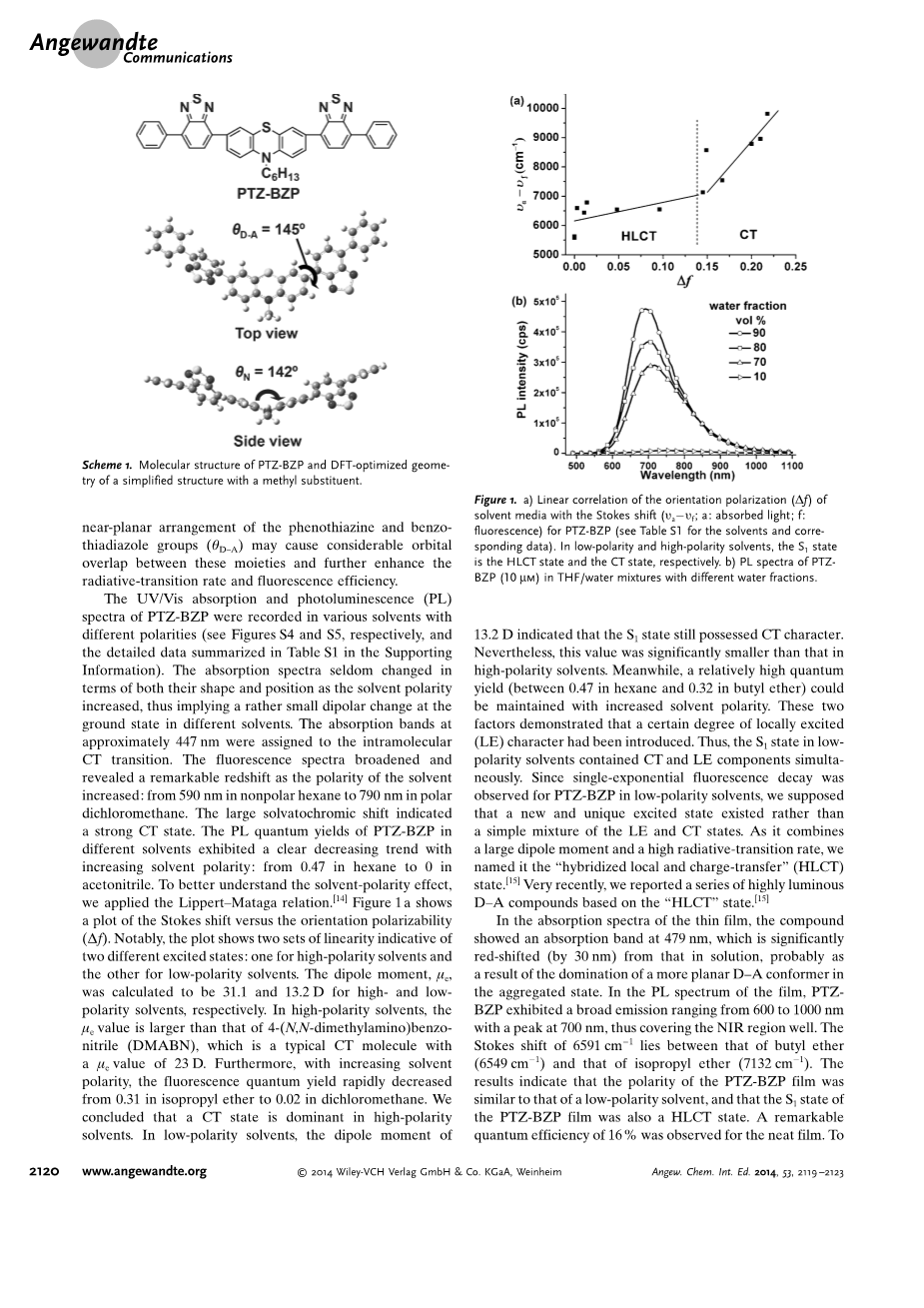
在不同极性的溶剂中测试了PTZ-苯并噻二唑的UV/Vis吸收光谱和光致发光光谱(PL)(分别见图S4和S5,详细数据见支撑资料表S1)。随着溶剂极性的增加,吸收光谱的形状和位置很少发生变化,这意味着在不同的溶剂中基态的偶极变化相当小。约447 nm处的吸收带归属于分子内CT跃迁。荧光光谱随着溶剂极性的增加而变宽,并显示出显著的红移:吸收带位置从在非极性正己烷的590 nm红移到在极性二氯甲烷的790 nm的溶剂变色位移表明了强的CT状态。不同溶剂中PTZ-BZP的PL量子产率随溶剂极性的增加呈明显的下降趋势:正己烷为0.47,乙腈为0。为了更好地理解溶剂极性效应,我们应用了Lippert-Mataga关系式[14]。1a显示了斯托克斯位移与取向极化率(Delta;f)的关系。值得注意的是,该图显示了两组线性关系两种不同的激发态:一种是高极性溶剂,另一种是低极性溶剂。偶极矩,mu;e,在高极性溶剂和低极性溶剂中分别为31.1和13.2 D。在高极性溶剂中,mu;e值大于4-(N,N-二甲氨基)苯并腈(DMABN),这是一种典型的CT分子,其mu;e值为23 D。此外,随着溶剂极性的增加,荧光量子产率迅速下降,在异丙醚中为0.31,在二氯甲烷中为0.02。我们的结论是,在高极性溶剂中CT状态占主导地位。在低极性溶剂中,偶极矩为13.2 D,表示S1态仍具有CT特征。然而,该值明显小于在高极性溶剂中的值。同时,随着溶剂极性的增加,可以保持较高的量子产率(在正己烷中为0.47,在丁基醚中为0.32)。这两个因素表明,体系中有一定程度的局部激发(LE)特性。因此,S1状态在低极性溶剂中同时含有CT和LE状态。由于观察到PTZ-BZP在低极性溶剂中的单指数荧光衰减,我们假设存在一种新的独特的激发态,而不是简单的LE和CT的混合态。由于它偶极矩大、辐射跃迁率高,我们将其命名为“局部杂化和电荷转移”(HLCT)态[15]。最近,我们报告了一系列基于“HLCT”态的高亮度D-A化合物[15]。
在薄膜的吸收光谱中,化合物的吸收带位于479 nm,并且该吸收带与在溶液中的吸收带相比明显红移(30 nm),这可能是聚集态中更为平面的D-A构象异构体占主导地位的结果。在薄膜的PL光谱中,PTZ-BZP表现出600~1000 nm的宽发射范围,在700 nm处有一个峰值,可以很好地覆盖近红外区域。斯托克斯位移为6591 cm-1,位于丁基醚(6549 cm-1)之间和异丙醚(7132 cm-1)。结果表明:PTZ-BZP薄膜的极性与低极性溶剂相似,PTZ-BZP薄膜的S1态也是HLCT态。观察到这种纯膜的显著量子效率为16%。为了研究聚合效应对荧光行为的影响,我们测量了PTZ-BZT在水组分不同的THF/水混合物中的PL光谱(fw;图1 b),可以直接调节溶剂极性和溶质聚集程度。fwle;60 vol%的溶液几乎不发射,PL曲线实际上是一条平行于x轴的平坦线,这主要是由于THF/水混合物的强极性所引起的CT效应。fwasymp;70%时荧光出现,并且荧光随fw的增加而增强。显然,PTZ-BZP具有聚集诱导发射(AIE)活性。[16] 产生AIE现象的主要原因可能是聚合态中的HLCT态比THF溶液中的CT态具有更有效的辐射电子跃迁。因此,HLCT态为高亮度D-A发色团的设计提供了一种新的方法。另一方面,分子间聚集体的形成往往限制了theta;N和theta;D-A的旋转,避免非辐射衰减通道的产生,提高了荧光效率[16]。
方案1 PTZ-BZP的分子结构和带有一个甲基取代基的简化结构的DFT优化几何构型
图 1 a) PTZ-BZP的溶剂介质取向极化(Delta;)与斯托克斯位移的线性关系(; a:吸收光; f:荧光) (溶剂和相应数据见表S1)。在低极性和高极性溶剂中,S1态分别为HLCT态和CT态。 b) PTZ- BZP(10 mm)在不同水组分的THF/水混合物中的PL谱。
为了验证PTZ-BZP作为光电子器件的高发光性能,我们制作了一种无掺杂的近红外OLED器件,其配置为:ITO/PEDOT : PSS(40 nm)/NPB(80 nm)/ TCTA(5 nm)/PTZ-BZP(30 nm)/TPBi(30 nm)/LiF(0.5 nm)/Al(120 nm;ITO = 氧化铟锡,PEDOT = 聚(3,4-乙基二氧硫代苯),PSS = 聚(苯乙烯磺酸盐),NPB = N,N´-双(萘-1-基)-N,N´-双(苯基)联苯胺,TCTA = 三(4-咔唑基-9-基苯基)胺,TPBi = 1,3,5-三(1-苯基-1H-苯并咪唑-2-基)苯。如图 2所示,PTZ-BZP器件的EL谱与蒸发薄膜的PL谱几乎相同。虽然我们的仪器(PR-650光谱仪)无法检测到780 nm以上的EL发光信号,但根据PTZ-BZP薄膜的PL谱可以推断EL光谱扩展到了近1000 nm。此外,在不同的驱动电压下,EL谱变化不大,说明PTZ-BZP器件具有良好的光谱稳定性。此外,基于PTZ-BZP的无掺杂近红外OLED最大EQE为1.54%,亮度为780 cdm-2。在300 mAcm-2的高电流密度下,PTZ-BZP器件的EQE仍然保持在1.17%的高水平,这表明效率下降相对较低。据我们所知,PTZ-BZP器件的性能使其成为最好的未掺杂近红外FOLEDs之一[6,8,11]。
辐射激子比的理论值由下式计算:
中是辐射激子比,是外量子效率,是光外耦合效率(约20%),是本征光致发光效率(约16%),是注入的空穴和电子的复合效率,理想情况下只有当空穴和电子完全平衡并完全复合形成激子时才是100%。因此,PTZ-BZP器件的值被计算为48%,这突破了传统FOLEDs的25%的辐射激子比的极限。由于在瞬态光致发光中没有观察到延迟荧光,并且电致发光的亮度随着电流密度的增加而线性增加,因此高辐射激子比似乎与不一些主流观点不一致,例如热激活延迟荧光(TADF)[12]或三重态—三重态湮灭(TTA)[17]。
图 2 器件的等电流密度特性。插图是EL光谱。
为了解释PTZ-BZP器件的高辐射激子比,我们根据M06-2X/6-31G(d,p)的TDDFT结果,计算和分析了单态和三态的能量分布和自然跃迁轨道(NTOs)(图3)。对于S1态来说,空穴和粒子的自然跃迁轨道在空间分离和轨道重叠之间表现出良好的平衡。分离良好的轨道使得CT态具有较大的偶极矩。另一方面,某些轨道重叠引起了LE特性,并保证了合理的辐射跃迁率。计算结果表明,CT态和LE态共存,符合我们对HLCT态的定义。低三重态T1是一个LE态,其空穴和粒子几乎完全重叠。S1态和T1态之间的能隙达到0.79 eV,相应的,TADF从T1态到S1态的逆系统间穿越(RISC)也不是一个容易的过程。T2和T1态是简并态。高三重态激发态T3(在本文中称为“热”激发态)被发现是一种HLCT态,其结构与S1态非常相似。如图3所示,S1态的能级(2.91 eV)和T3态的能级(2.92 eV)几乎相同。这种小的但单重态—三重态分裂可以提供一个潜在的RISC T3→S1过程,我们称之为“热激子”过程[18]。另一方面,T3态和T2或T1态的能隙为0.76 eV。根据能隙定律,从T3态到T2态的内转换速率可能低于T3态到S1态的RISC速率(kRISC)[19]。此外,有报道称,在一些含有硫原子的杂环体系中,由于自旋轨道耦合的改善,RISC速率可以大大提高[20]。因此,当三重态激子弛豫到T1的最低振动能级时,其中的一部分可以通过T3→S1通道转换成单线态激子。因为三重态态激子的浓度是动态平衡,远高于单态激子,在电致发光过程中,动态平衡会促进三重态激子向单态激子转化。我们推断,热激子过程提供的高效RISC是PTZ-BZP器件中辐射激子比超过25%的原因。
图 3 a) S1, TI, T3的自然跃迁轨道 b) 单重态和三重激发态的能量图 c) EL过程中的激子弛豫模型。RISC:统间反向交叉; IC(T):三重态
剩余内容已隐藏,支付完成后下载完整资料

英语原文共 5 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[271202],资料为PDF文档或Word文档,PDF文档可免费转换为Word

