
英语原文共 3 页,剩余内容已隐藏,支付完成后下载完整资料
窗体顶端
通过电氧化合成新的吩噻嗪的一锅,简单和清洁的方法的氢醌在2-氨基苯硫酚存在下
Omid Ghaderi,Alireza Asghari,* Mohsen Ameri,以及Maryam RajabiSemnan大学化学系,Semnan 35195-363,伊朗
(E-mail: aasghari@semnan.ac.ir)
摘要:在2-氨基苯硫酚(3)的存在下,通过电化学氧化氢醌(1a和1b)在含有0.15mol·L-1磷酸盐(pH6.0)的水/乙醇(90/10))溶液中进行新的吩噻嗪衍生物的电化学合成, 循环伏安法(CV)和受控电位库仑法(CPC)方法。 结果表明,电化学产生的对苯醌通过ECEC机制与3分子在1,4-迈克尔加成反应中分离,以良好的收率和高纯度形成吩噻嗪衍生物6a和6b,使用简单的一锅清洁, 和非分解电池中的三个碳电极的非催化剂方法。
关键词:吩噻嗪、电化学氧化、对苯二酚
正文:
吩噻嗪是含有通过氮和硫原子在三环体系中连接的两个苯环的杂环分子。近几十年来,由于各种生物活性如镇静剂,抗炎药,抗疟药,抗精神病药,抗菌药,抗结核药,抗肿瘤药,抗组胺药,镇痛药等,因此大量关注吩噻嗪衍生物的合成。已经观察到一些吩噻嗪抑制包括人免疫缺陷病毒(HIV)在内的病毒的细胞内复制.7另一方面,已经报道了这些衍生物中的一些具有显着的抗癌活性.8由于这些杂合物的重要性增加在过去几年中,在新一代发挥其生物活性的10H-吩噻嗪方面进行了尝试。氢醌氧化还原体系由于其生物和环境重要性以及其氧化还原化学的经典基本特征而得到广泛研究.9这些化合物可以电化学氧化成对醌。形成的对醌具有相当的反应性,可以被不同的亲核试剂攻击。迄今为止,在不同的亲核试剂存在下,氢醌已被用作1,4-迈克尔受体.122根据吩噻嗪衍生物的生物学和药物性质,绿色化学在有机合成中的重要性日益增加,需要开发在化学转化的更有效和环境友好的过程中,13在温和条件下,在存在和不存在2-氨基苯硫酚(3)作为亲核试剂的情况下,已经研究了氢醌和2,3-二甲基氢醌1a和1b的电化学氧化(无热,无化学试剂,无压力,无任何催化剂)在与乙醇混合的水性介质(磷酸盐溶液)中。这导致在使用三个石墨电极的未分隔的电池中使用容易,一锅和环保(无试剂,无催化剂,无任何有毒溶剂)电化学方法合成新的吩噻嗪衍生物。
窗体顶端
反应设备按照支持信息和早期文件中所述使用.12,14所有化学材料均购自默克(德国达姆施塔特)。 这些化学品无需进一步纯化即可使用。
窗体底端
在典型的方法中,加入100毫升磷酸盐溶液(c = 0.15mol L11,pH6.0 /乙醇(90/10 v:v))0.5mmol氢醌1a和1b以及0.5mmol 2-氨基苯硫酚(3)在0.5V下相对于Ag / AgCl电解(KCl,3M)通过控制电位库仑法与三碳电极在未分割的电池中。当电流降低超过95%时,电解结束,同时进行反应,该过程重复六次。为了重新活化碳阳极,在电解过程中用丙酮洗涤它们。在电解结束时,通过离心收集沉淀物。产物在温热的乙醇中结晶。纯化后,产物的特征是熔点,FTIR,1 H NMR,13 C NMR,和元素分析(CHN)。化合物6a:产率:70%; Mpgt; 250℃(分解); FT-IR(KBr,cm -1):3400(宽,OH),3200(宽,NH); 1 H NMR(400MHz,CDCl 3):5.29(s,NH),6.66(d,1H,芳族,J = 8.6Hz),6.82(d,1H,芳族,J = 8.0Hz),7.657.69(m, 4H,芳族),9.3(宽峰,2H,OH); 13 C NMR(100MHz,CDCl 3)147.5,151.3。肛门。计算。对于C 12 H 9 NO 2 S:C,62.32; H,3.92; N,6.06; O,13.84%。实测值:C,62.56; H,3.86; N,6.12; O,13.71%。化合物6b:产率:71%; Mpgt; 250℃(分解); FT-IR(KBr,cm -1):3380(宽,OH),3220(宽,NH)。 1 H NMR(400MHz,CDCl 3):2.19(s,3H,甲基),2.23(s,3H,甲基),5.24(s,NH),7.637.80(m,4H,芳族),9.15(宽峰,2H, ); 13C NMR (100 MHz, CDCl3):curren;14.5,15.1,76.7,77.1,77.4,115.5,125.4,125.4,125.9,127.5,127.9,127.9,131.8,133.6,135.5,148.7,150.5。肛门。计算。对于C 14 H 13 NO 2 S:C,64.84; H,5.05; N,5.40; O,12.34%。实测值:C,64.76; H,5.23; N,5.36; O,12.71%。
窗体顶端
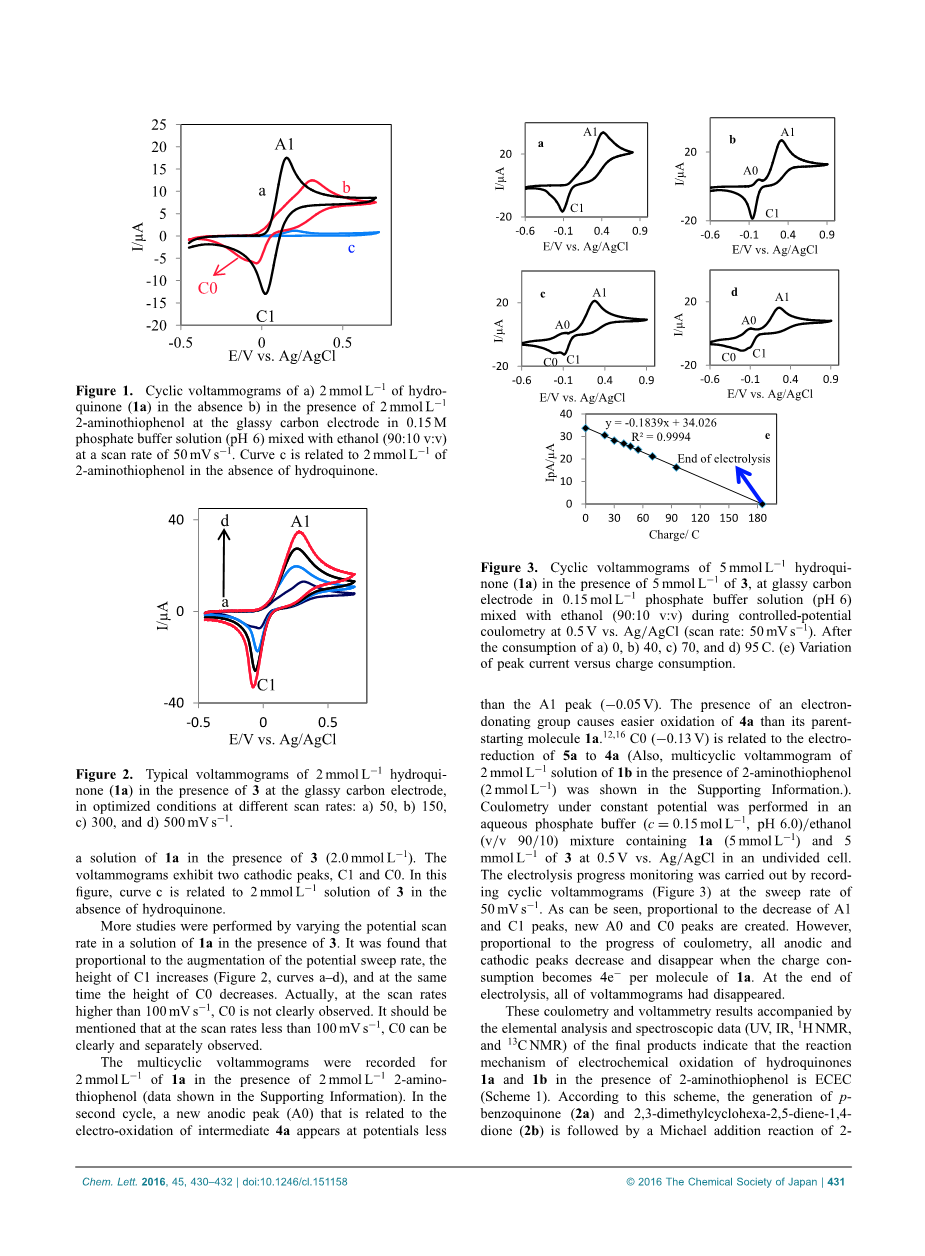
图1显示了2.0 mmol L 1氢醌(1a)在含有0.15mol·L-1磷酸盐(pH 6.0)的水/乙醇(90/10)溶液中的循环伏安图,曲线a。 可以看出,获得了一个阳极(A1)和阴极峰(C1)。 阳极和阴极峰是对应的,并且对应于1a到对苯醌(2a)的转变,反之亦然,在准可逆双电子过程中。 循环伏安图中的峰值电流比(IpC1 / IpA1)约为1。 因此,可以得出结论,在实验条件下在电极表面产生的对苯醌是稳定的,并且任何副反应如羟基化和/或二聚反应都太慢,不能在CV时间刻度上观察到15。 获得对氢醌(1a)或1b的电化学氧化的进一步支持,在作为亲核试剂的2-氨基苯硫酚(3)的存在下研究反应。 图1,曲线b,显示了第一个获得的CV
窗体底端
图1 a)2mmol L 1氢醌(1a)的循环伏安图,在不存在b)在2mmol L 1 2-氨基苯硫酚存在下,在0.15M磷酸盐溶液(pH6)的玻璃碳电极中混合氢醌 乙醇(90:10 v:v),扫描速率为50mV s为1.在不存在氢醌的情况下,曲线c与2mmol L 1的2-氨基苯硫酚有关。
图2.在玻璃碳电极存在下,在3个玻璃碳电极存在下,2 mmol L 1 hydroqui-none(1a)的典型伏安图,在不同扫描速率的优化条件下:a)50,b)150,c)300,d) 500 mV s1。
在3(2.0mmol L 11)存在下,将1a的溶液。 伏安图显示了两个阴极峰C1和C0。 在该图中,曲线c与不存在氢醌的2mmol L 1溶液3相关。
通过在3的存在下改变1a的溶液中的潜在扫描速率来进行更多研究。发现与潜在扫描速率的增加成比例,C1的高度增加(图2,曲线ad),并且在 同时C0的高度减小。 实际上,在扫描速率高于100 mV s1的情况下,C0没有明确的观察。 应该提到的是,在扫描速率小于100mV s1的情况下,可以清楚和分别地观察到C0。
在2 mmol L 1 2-氨基 - 苯硫酚的存在下,记录2 mmol L11的1a多环伏安图(数据显示在支持信息中)。 在第二个循环中,与中间体4a的电氧化相关的新的阳极峰(A0)出现在较小的电位
图3 5 mmol L 1氢化喹啉的循环伏安图没有(1a)在玻璃碳下存在5 mmol L 1的3电极在0.15 mol Ll1磷酸盐溶液(pH 6)在0.5V对Ag / AgCl的控制电位库仑法(扫描速率:50mV s1)下与乙醇(90:10 v:v)混合。 消耗a)0,b)40,c)70和d)95 C.(e)峰值电流与电荷消耗的变化。
比A1峰值(sup1;0.05V)。 给电子基团的存在导致4a比其母体起始分子1a.12.16更容易氧化C0(0.103V)与5a至4a的电还原有关(另外,2mmol的多环伏安图 在支持信息中显示了1b在2-氨基苯硫酚(2mmol L 1)存在下的L11溶液。 在磷酸盐水溶液(c = 0.15mol L11,pH6.0)/乙醇中进行恒定电位的库仑法
(v / v 90/10)混合物,其含有1a(5mmol L11)和5在未分裂的细胞中,在0.5V下相对于Ag / AgCl的mmol L11为3。电解进度监测通过记录循环伏安图(图3),扫描速率为50mV s1进行。 可以看出,与A1和C1峰值的降低成比例,产生新的A0和C0峰。 然而,与电量计算的进展成比例,当电荷消失成为每分子1a的4e 1时,所有阳极和阴极峰都减小并消失。 在电解结束时,所有的伏安图都消失了。
这些库仑法和伏安法结果伴随着最终产物的元素分析和光谱数据(UV,IR,1H NMR和13C NMR)表明,在2-氨基苯硫酚存在下氢醌1a和1b的电化学氧化反应机理是 ECEC(方案1)。 根据该方案,生成对苯醌(2a)和2,3-二甲基环己-2,5-二烯-1,4-二酮(2b)之后,进行迈克尔加成反应,
氨基苯硫酚(3),其生成中间体4a和4b。 这些化合物4a和4b的氧化比借助于给电子基团的存在使母体起始分子1a和1b的氧化更容易.17第二对电子的抽象导致形成5a和5B。 然后,源于胺基(NH 2)对苯醌片段的亲核攻击和随后的环化的分子内加成反应产生吩噻嗪衍生物6a和6b。 由于产物在磷酸盐溶液介质中的不溶性,在制备反应期间避免了6a和6b的过氧化。 反面上的阴极反应是氢的释放。
在3〜9的各种pH值下,研究了pH对电合成和苯并噻嗪衍生物产率的影响。最佳结果表明,纯度为6和6的纯产物的最大量为6(表1)。
总之,我们使用CV和可控电位库仑法,从氢醌1a和1b与2-氨基苯硫酚(3)的反应中成功地进行了吩噻嗪6a和6b的电化学合成。结果表明氢醌1a和1b被氧化成它们各自的对苯醌2a和2b。形成的对苯醌受到3的攻击,导致中间体4a和4b。然后中间体被氧化并进行分子内迈克尔加成反应和随后的环化,这导致产物的产生。得到的产物收率高,纯度高。我们的研究已经导致了在温和的反应条件下发生的一种简单,环保的方法。此外,在水/乙醇混合物中合成有价值的化合物,其中乙醇与水的比例小,原子经济性高,一步法,并且使用电极作为电子源代替有毒试剂,是本研究的优点它引入电化学作为合成新的杂环化合物如吩噻嗪的“有力工具”。
支持信息可从http://dx.doi.org/ 10.1246 / cl.151158获得。
参考文献:
[1]M. K. El-Said, Pharmazie 1981, 36, 678.
[2]T. Ram, R. Tyagi, B. Goel, K. Saxena, V. Srivastava, A. Kumar, Indian Drugs 1998, 35, 216.
[3]J. N. Domiacute;nguez, S. Loacute;pez, J. Charris, L. Iarruso, G. Lobo,A.Semenov, J. E. Olson, P. J. Rosenthal, J. Med. Chem. 1997, 40, 2726.
[4]J. P. Raval, K. R. Desai, ARKIVOC 2005, Part xiii, 21.
[5]M. Viveiros, L. Amaral, Int. J. Antimicrob. Agents 2001, 17, 225.
[6]N. Motohashi, M. Kawase, S. Saito, H. Sakagami, Curr. Drug Targets 2000, 1, 237.
[7]Y. P. Singh, A. S. Tomar, S. K. Vijay, Asian J. Spectrosc. 2010, 14, 57.
[8]T. Kurihara, K. Nojima, H. Sakagami, N. Motohashi, J. Molnaacute;r, Anticancer Res. 1999, 19, 3895.
[9]M. A. Ghanem, Electrochem. Commun. 2007, 9, 2501.
[10]M. Ameri, A. Asghari, A. Amoozadeh, D. Nematollahi,M.A. Chamjangali, L. Boutorabi, Prog. React. Kinet. Mech. 2014, 39, 391.
[11]M. Ameri, A. Asghari, A. Amoozadeh, H. Daneshinejad, D. Nematollahi, Chin. Chem. Lett. 2014, 25, 797.
[12]M. Ameri, A. Asghari, A. Amoozadeh, M. Bakherad, D. Nematollahi, J. Electrochem. Soc. 2014, 161, G75.
[13]P. T. Anastas, M. M. Kirchhoff, Acc. Chem. Res. 2002, 35, 686.
[14]A. Asghari, M. Ameri, A. A. Ziarati, S. Radmannia, A. Amoozadeh, B. Barfi, L. Boutorabi, Chin. Chem. Lett. 2015, 26, 681.
[15]T. E. Young, J. R. Griswold, M. H. Hulbert, J. Org. Chem. 1974, 39, 1980.
[16]M. Ameri, A. Asghari, A. Amoozadeh, M.
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[28542],资料为PDF文档或Word文档,PDF文档可免费转换为Word
课题毕业论文、外文翻译、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。

