
英语原文共 9 页,剩余内容已隐藏,支付完成后下载完整资料
比较消化物和化肥对土壤细菌的影响
Melanie Sapp, Mark Harrison, Ummey Hany, Adrian Charlton, Richard Thwaites
摘要:不同的施肥制度,即在农业中使用无机或有机肥料,被认为会对土壤细菌产生不同的影响。本研究采用温室试验,通过16S rRNA基因v1-v3区的热测序,研究了添加无机肥料或污泥消化物对土壤细菌群落结构和多样性的影响。以春小麦(小麦品种Paragon)为模型作物,监测其生长(以总干重计)及128天以上3个时间点土壤氮磷变化。总的来说,40种细菌以变形杆菌、酸杆菌和放线菌为主。此外,在所有样本中均发现类杆菌、双子体、氯仿和ws3。在所有样品中都发现了平生菌、厚壁菌、亚硝基螺旋菌、候选菌群SPAM和犰狳科的成员,但含量较低。在蛋白菌门内,alpha;和beta;蛋白菌是最常见的。在整个实验过程中,我们观察到了放线菌、变形菌和嗜酸杆菌的主要处理差异。在整个实验过程中,与平生菌相关的序列(与氮循环有关)在所有处理中都有所下降。统计分析表明,主要的营养添加和植物生长影响了细菌群落结构。处理本身的影响可归因于小麦生长的不同收获,特别是在试验结束时比较群落时。结果表明,不同肥料的施用不仅直接影响细菌群落,而且间接影响细菌群落。
关键词:土壤微生物群落 消化物 春小麦 焦磷酸测序 温室实验
- 介绍
人口增长、耕地可用性下降、土地利用变化以及气候变化导致的不可预测的天气状况,都增加了农业生产力的压力(Banwart,2011)。生产力反过来又与土壤微生物群落密切相关,因为通过土壤微生物、营养循环、植物生长和疾病抑制之间的密切关系,在保持健康土壤方面构成了基本部分(Garbeva.et.al,2004;van der Heijden.et.al,2007;Maron.et.al,2011)。然而,目前的耕作方式主要影响土壤中的微生物多样性,导致其减少(Roesch.et.al,2007)。具体来说,在轮作中观察到细菌数量减少,而某些作物的持续种植导致群落结构的变化(Ng.et.al,2012)。据认为,目前使用的无机肥料是对内在土壤微生物群落的一种主要有害影响,并已显示出导致英国所有土地利用类型的土壤有机质下降(Kibblewhite.et.al,2008)(Bellamy.et.al,2005)。具体来说,添加氮会影响碳转化(KibbleWhite.et.al,2008),从而对微生物群落的功能产生不利影响,对特定功能可能产生相当大的影响。一个例子是甲烷代谢:已知农业土壤中甲烷营养因子的多样性较低(Levine.et.al,2011)。
最近,人们认为将消化物用作土壤添加剂是有益的(Alburquerque.et.al,2012),因为它含有磷等必需营养素。尤其是磷是一种矿物和减少的资源(Malik.et.al,2012),生产在原材料运输和开采中需要巨大的环境成本。消化物是通过废物的厌氧降解产生的,主要来源于食物、作物(无论是专用种植还是作物残留物)或废水处理过程中产生的污泥,其中的固体部分可能含有丰富的营养素,包括氮和磷以及纤维素等有机物。
尽管各国已制定了控制重金属和病原体(Paavola和Rintala,2008;BSI,2010)和有机污染物(Al Seadi,2009)的标准,以尽量减少消化物的危害,但忽略了使用消化物对土壤的其他潜在影响。其中包括土壤有机质组成的修正、营养物质(例如氮化合物)超载以及微生物群落组成的变化(Alburquerque.et.al,2012;Tambone.et.al,2009;Fuchs.et.al,2008)。即使在废水处理到半干旱草原的生物固体添加率相当低的情况下,也可以观察到后者(Sullivan.et.al,2006)。尤其是,当污水污泥被添加到长期的土壤有机物实验中(B_rjesson.et.al,2012)时,革兰氏阳性细菌变得更为丰富,同时,与污泥重金属输入相关的微生物群落结构的总体变化也很大。此外,制革厂污泥导致了强烈的群落和生理变化(Nakatani.et.al,2011)。因此,对污泥来源的考虑非常重要。这一点得到了微生物对仅使用有机肥料的不同反应的支持,这导致了对细菌和真菌的不同反应,有利于革兰氏阴性细菌和真菌群落。相比之下,对革兰氏阳性细菌(如放线菌)观察到负作用(Zhang.et.al,2012)
除微生物多样性外,关于消化物对用于作物生产的土壤条件的影响的实验需要纳入作物生产力的测量。这些研究发现,虽然可以使用非生物肥料或消化物获得类似的作物产量(Haraldsen.et.al,2011),但添加肥料和生物肥料的组合导致微生物生物量、碳和氮矿化、土壤呼吸和酶活性(Dinesh.et.al,2010)以及细菌增加。l多样性(Gu.et.al,2009)。然而,仅添加非生物营养素就导致了土壤微生物群落的压力(Dinesh.et.al,2010)。在我们的研究中,我们测试了植物生长和土壤细菌多样性与添加消化或非生物肥料的关系。营养状况是基于英国农民最近报告使用的非生物肥料(defra,2010)和英国环境署(2010)关于使用消化物的建议,使用的氮素水平(Abubaker et al., 2013)与大多数对植物生物量的重要贡献者相似。
我们假设不同的营养来源(即有机肥料和无机肥料)对土壤微生物群落和植物生长都有显著影响。植物干重,作为生长的一个衡量指标,与土壤细菌群落的变化一起测量。到目前为止,只有少数研究使用分子工具比较了传统农业技术或使用消化物作为肥料后土壤微生物群落的变化。我们已经应用了高温测序,因为这项技术有可能揭示探索不足的微生物多样性(Maron.et.al,2011),这可能对在不断变化的条件下适应社区发挥关键作用。更具体地说,我们以春小麦(小麦品种Paragon)为模型作物,研究了不同营养添加剂(即污水污泥和无机肥料产生的消化物)对土壤细菌群落结构的影响。在试验开始时,以植物生物量为因子,对主要营养素N、P和细菌群落组成进行了评价,并在65、128天后进行了重复试验。
- 材料与方法
2.1实验设计
建立了一个完全随机分块设计的温室实验,包括三种处理方法,每种方法重复两次。在0.025立方米的罐子中填充一种农业土壤,称为砂岩高地,具有已知的农药历史。后者于201010月从英国利明顿温泉(Lat=52.32939,Long=1.49949)采集,深度为0-20厘米。五来,土壤一直作为备用(无作物种植)进行管理,并收到少量草甘膦。
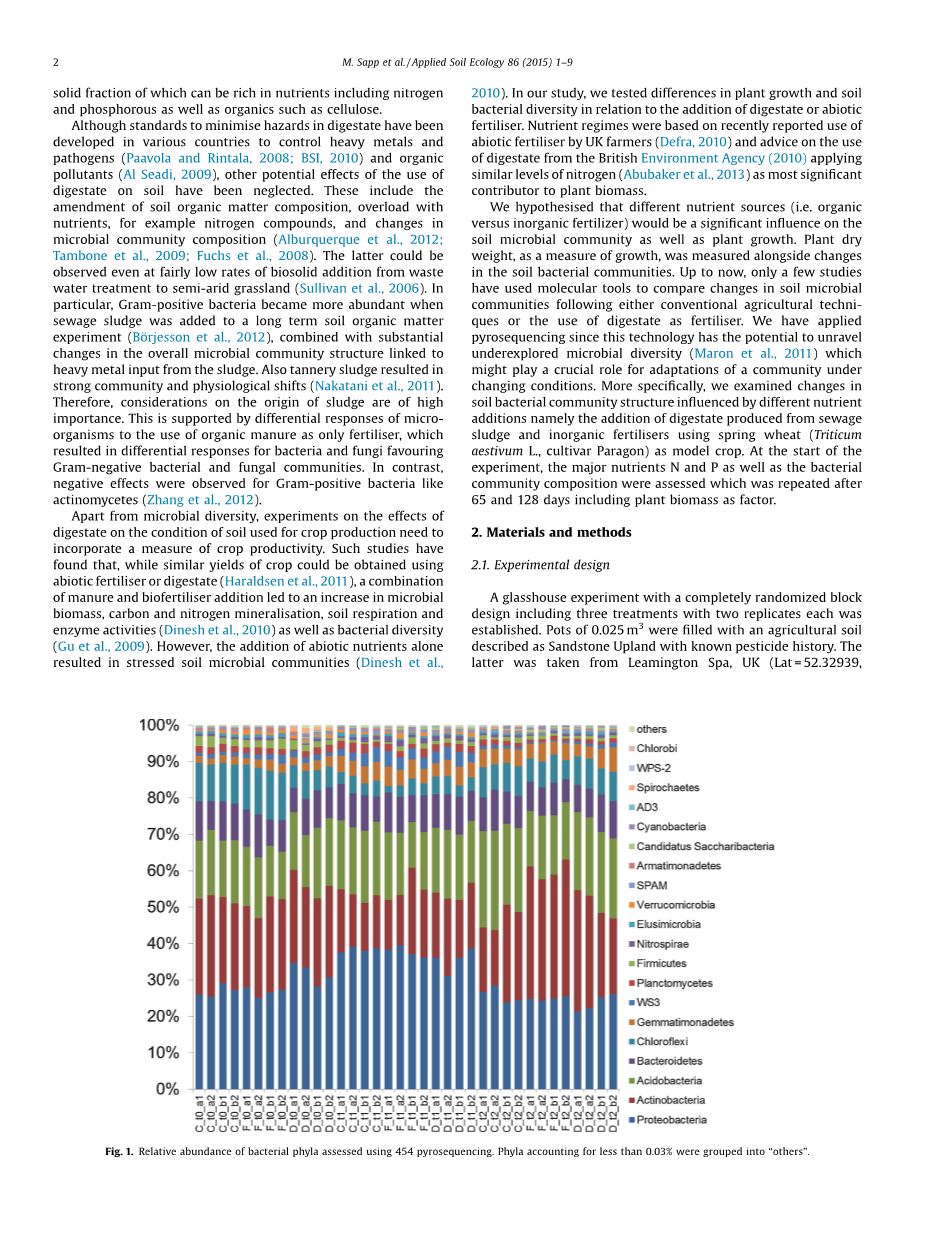
图1 用454热测序法测定细菌门的相对丰度(小于0.03%被归为“其他”)
总的来说,在3个不同的时间点(20112月21日t0,20114月26日t1,20116月27日t2)进行破坏性采样,共设置对照、施肥和添加消化物三种处理,一式两份。除了对照之外,还添加了新鲜消化物或无机肥料来模拟当前使用的无机肥料(defra,2010)和英国指南(环境署,2010)建议的消化物。添加相当于1440 mg N和0.0343 mg P的特殊新鲜消化物,而化肥处理则接受相当于1440 mg N和480 mg P的氮和磷,模拟当前147 kg ha-1 N和45 kg ha-1 P的做法,将各自的浓度施加于土壤表面并静置24 h。在试验中,试验结果表明,试验结果与试验结果一致,对照样品加入等量的水。彼得斯专业高硝基水溶性肥料被用作无机肥料(英国约克州东骑园艺公司),20112月从英国约克州纳伯恩的废水处理厂(约克郡水)采集了新鲜消化物。纳伯恩厌氧消化池接收城市污水污泥,在单级反应器中进行消化。随后对包含时间点t0的这些盆进行次采样,而其余12个盆则随机放置在温室中,应用“随机序列生成器”(www.random.org)算法确定位置。从Wynnstay Group plc(Llansantffraid,UK)获得的18个品种Paragon T.L.种子在每罐中4行显示,每行每个种子之间6.6 cm,第一排种子距盆子边缘6.5厘米。接下来的两行间隔7厘米,每行含有5粒种子。最后一排离第三排7厘米,离盆子边缘6.5厘米,由四粒种子组成,温室内的温度保持在21○C,没有使用人造光,因此日出和日落时间符合英国的夏季条件。根据温室外的每日测量,在生长期间的平均光强度记录为134.7 W m-2。
在分析之前,土壤在黑暗中在室温下风干1周。筛选2 mm后,进行化学分析。用于分子分析的样品冷冻在-20○C。
表1 16S rRNA基因序列综述以及3%序列距离下簇的多样性和丰富度估计
2.2作物分析
每种作物都被收割,根茎被移除。将每个罐中的作物捆绑在一起,在80○C下干燥24小时。从烘箱中取出干燥的作物后,在干燥剂中冷却至环境温度,然后测量干重。由西约克郡分析机构(英国莫利)使用Dumas方法测量氮含量,其中样品在氧气中燃烧。然后用热导检测器测量二氧化氮。将总磷(P)测量为Olsens P。详细地说,将100 ml新制备的碳酸氢钠试剂(0.494 M碳酸氢钠,0.05%mv-1丙烯酰胺,ph 8.5)添加到50 g土壤中,摇晃30分钟,之后,溶液通过Whatman 2号过滤器(英国麦德斯通GE Healthcare)。最初的几毫升过滤液被丢弃,剩下的液体被收集起来。采用紫外分光光度法进行分析。为此,制备了钼酸铵试剂(9.7 mM钼酸铵,0.45 mM酒石酸钾抗蚀剂,14.5%硫酸),并在黑暗中4○C下储存。以Olsen试剂为稀释剂,从20 mg L-1储备液中制备0-7 mg P kg-1的口径。将等量(167 ml)的校准剂或样品用移液管移入2 ml eppendorf管中,依次添加33 ml 1.5 m硫酸、667 ml 0.15%mv-1钼酸铵试剂和167 ml新制备的1.5%mv-1抗坏血酸溶液。将溶液混合,静置30分钟,然后将其转移到石英试管中,在880 nm处测量吸光度。
图2 A:基于相对细菌丰度的布雷-柯蒂斯相似性的NMDS双区(对照(sect;)、消化物(“)和肥料(*)处理的样品,(t0–t2)表示时间点) B:各时间点的复制相互重叠,基于细菌相对丰度的CCA双肽(对照(sect;)、消化物(“)和肥料(*)处理的样品,(t0–t2)表示时间点,字母表示重复组,数字表示每个组的子样本。影响因子为氮(N)、磷(P)、时间和植株干重)
2.3微生物分析
对于每个时间点,通过从每个时间点(1和2)的不同侧面采集两个独立的子样本,对重复的时间点(A或B)进行采样,对这些样本分别进行DNA提取、扩增和测序。
-
-
- 16S rRNA基因v1–v3区的DNA提取和热测序2.3.1.16S rRNA基因v1–v3区的DNA提取和热测序
-
根据制造商的方案,使用Powersoil1DNA分离试剂盒(Mo Bio,Carlsbad,USA)从0.25 mg土壤中提取DNA。根据16S rRNA基因的v1–v3区域评估细菌群落结构(Hamady.et.al,2008;Turner.et.al,1999)。PCR引物适用于454个扩增序列,其中25-mer lib-l特异性序列适配器(带下划线)后接454个扩增序列特异性4-mer扩增键(斜体),该键与10-mer条形码序列一起用于辅助复用(nnnn)和模板特异性引物(粗体)序列, 16s_27F(50-CCATTCTCCCTGCGTCCGACTCAGNNAGAGTT-GATCMTGCTCAG-30;Hamady.et.al,2008)和16s_519r (50-CCTATCCCCTGTGCCTTGGCAGTCGGWATT-TACCCGGCKGCTG-30(Turner.et.al,1999)。表S1给出了使用的10-mer条形码的序列。使用KAPA HiFi PCR试剂盒进行25 ml的扩增反应(KAPA Biosystems, Wilmington, USA),含有5毫升KAPA气相色谱缓冲液,每个引物0.3 mm,dNTPs 0.3 mm,KAPA HiFi DNA聚合酶0.5 U,模板DNA 40 ng。最终反应体积由无核酸酶水组成(Severn Biotech, Kidderminster, UK)。在C1000热循环器(Biorad, Hemel, Hempstead, UK)上进行放大,从95○C初始单一变性步骤开始4分钟,然后在94○C下变性35个循环20秒,在57○C下退火15秒,在72○C下延长30秒,然后在72○C下最终延长5分钟。PCR产物在2%琼脂糖凝胶上进行检查,并在紫外线下进行可视化。使用Quavik凝胶提取试剂盒(Qiagen, Hilden, 德国),在UV下尽可能快地从凝胶中分离出单独的PCR产物。凝胶滴样PCR产物经纳米滴度定量,共检测到十二个PCR产物。使用Agencount AM Pure XP(Beckman Coulter, Brea, USA)进一步纯化聚合的pcr产物,然后使用Roche的建议制备一个扩增子库。在454基因组分析仪(Roche, Burgess Hill, UK)上使用GS-FLX钛化学在两次单独运行中进行测序,其中一次运行的时间点为t0和t1(SRR1378331),另一次运行的时间点为t2样品(SRR1378333)。
-
-
- 数据分析
-
Qiime Pipeline v.1.5.0(Caporaso.
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[21085],资料为PDF文档或Word文档,PDF文档可免费转换为Word
课题毕业论文、外文翻译、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 太阳辐射减弱对微生物多样性的影响开题报告
- 在斯里兰卡副湿地中大气二氧化碳对于水稻(Oryza sativa)的光合作用和水分关系的升高反应外文翻译资料
- 比较消化物和化肥对土壤细菌的影响外文翻译资料
- 五年臭氧熏蒸下臭氧浓度升高可提高小麦根际的硝化和反硝化酶活性外文翻译资料
- 深层植被去除促进了富营养化湖泊中浮游植物功能群的转移外文翻译资料
- 含钠农田条件下渍水对不同品种冬小麦物质含量、生长和产量的影响外文翻译资料
- 模拟酸雨对中国北部地区冬小麦(黑麦)的生长影响外文翻译资料
- 生物膜及其与生物炭联合法在砷去除中的应用外文翻译资料
- 在Richardson模型中引入季节参数变化估算降雨侵蚀力外文翻译资料
- 在南多布罗加种植秋小麦的农业气象条件外文翻译资料

