
英语原文共 7 页,剩余内容已隐藏,支付完成后下载完整资料
摘要:卤化钙钙钛矿太阳能电池是最近的一个突破性发展,它实现了电力转换效率超过18%。由于AMX3钙钛矿的显着特性,这可能使其具有独特的半导体性能。最有效的太阳能电池利用了CH3NH3PbI3钙钛矿,其带隙Eg为1.55eV。然而,如同在CH3NH3SnI3(Eg = 1.3eV)的情况下,钙钛矿的带隙可以在近红外区域大大提高效率。进一步改进的一个显著的方法来自于CH3NH3Sn1-xPbxI3固溶体,其显示了带隙演变的异常趋势,相比于最低的末端成员CH3NH3SnI3,接近x = 0.5的成分显示较低的带隙(Egasymp;1.1eV)。这里我们使用第一性原理计算来证明自旋轨道耦合(SOC)和晶格畸变之间的竞争是CH3NH3Sn1-xPbxI3中带隙的异常行为引起的。 随着x增加,SOC导致线性减小,而晶格畸变是由于在x = 0.5附近的组成诱导相变引起非线性增加导致的。我们的研究结果表明,电子结构工程可以在优化光伏性能方面发挥关键作用。
由于再生能源广泛的应用价值,新型光伏材料的研究与开发迅速增长。尤其是基于钙钛矿的AMX3(A =小有机阳离子; M = Pb,Sn和X = I,Br)太阳能电池是由于其具有非常高的效率和易于制造固态太阳能电池的潜力,而使得该领域发生了变革。CH3NH3PbI3应用于太阳能电池吸收体,具有优异的材料特性,如高迁移率,长扩散长度,大光学吸收和合适的带隙(Eg = 1.55eV)。 通过连续不断的实验,在卤化钙钙钛矿太阳能电池的性能方面取得了显著增强。将太阳能吸收范围扩大到近红外光谱可进一步提高卤化钙钙钛矿太阳能电池的性能。考虑到太阳光谱的性质,可以在带隙为Eg = 1.1 eV的单结器件中实现最大理论效率。 在最近的研究进展中,在混合卤化钙钙钛矿体系CH3NH3Sn1-xPbxI3中已经实现了这种策略,表明通过在钙钛矿的金属部位合金化Sn和Pb原子,带隙可以降低到Eg = 1.17eV。然而,异常的潜在机制仍然很少了解,但可以将其与MA1-xFAxPbI3中观察到的相似效应进行比较,其中阳离子尺寸间接决定了钙钛矿的带隙。 有趣的是,Ogomi等报道了类似系统中的单调行为,这与非单调带隙行为相矛盾。 因此,需要对非单调行为的基本原理进行深入的研究以清楚地理解系统。
合金化合物中的非单调带隙行为是半导体领域的根本性质。 通常,合金化合物A1-xBx与x的带隙展开式很好地由正常数b表示,具有以下关系。
这被称为带隙弯曲,所谓的弯曲参数b给出了与简单线性插值的偏差。b的值通常较小且与组分无关,而在大多数情况下,b的小值可以通过无序效应来解释。相反,弓形参数在某些化合物中变得巨大或成分依赖。在这种情况下,带隙弯曲的起点通常更复杂。例如,GaAs1-xNx具有巨大的弯曲参数(体积为16eV)和几个物理起源如两级抗锯齿,带隙内的N杂质带形成,或导带内的内部混合和与N群集的相互作用。同样,CH3NH3Sn1-xPbxI3固体溶液的带隙行为绘制出一种高度非线性和非单调曲线,这不是通过具有常数b的简单弯曲来解释的,因此需要更精确的电子结构计算进行更深入的理论研究,以揭示其潜在的机制。
我们显示CH3NH3Sn1-xPbxI3固溶体中异常带隙减少的起源是由强自旋轨道耦合(SOC)引起的带隙变窄的拮抗作用和由x依赖的结构失真引起的带隙扩大钙钛矿网。第一原理电子结构计算显示,SOC引起带宽随着x线性增加的带隙减小,而由相变(从x4gt; P4mm到I4cm)导致的晶格失真会导致与微妙的几何变化相关的带隙增加。在相变之前和之后的带隙趋势的这种差异解释了在x = 0.5附近观察到的带隙行为的实质变化。它还解释了在 0.1 lt;x lt;0.5 的范围内在CH3NH3Sn1-xPbxI3固溶体中存在1.1eV的意想不到的最小带隙的存在。这种效应导致将太阳吸收光谱扩展到混合Pb / Sn卤化物钙钛矿及其太阳能电池中的较低能量的能力。
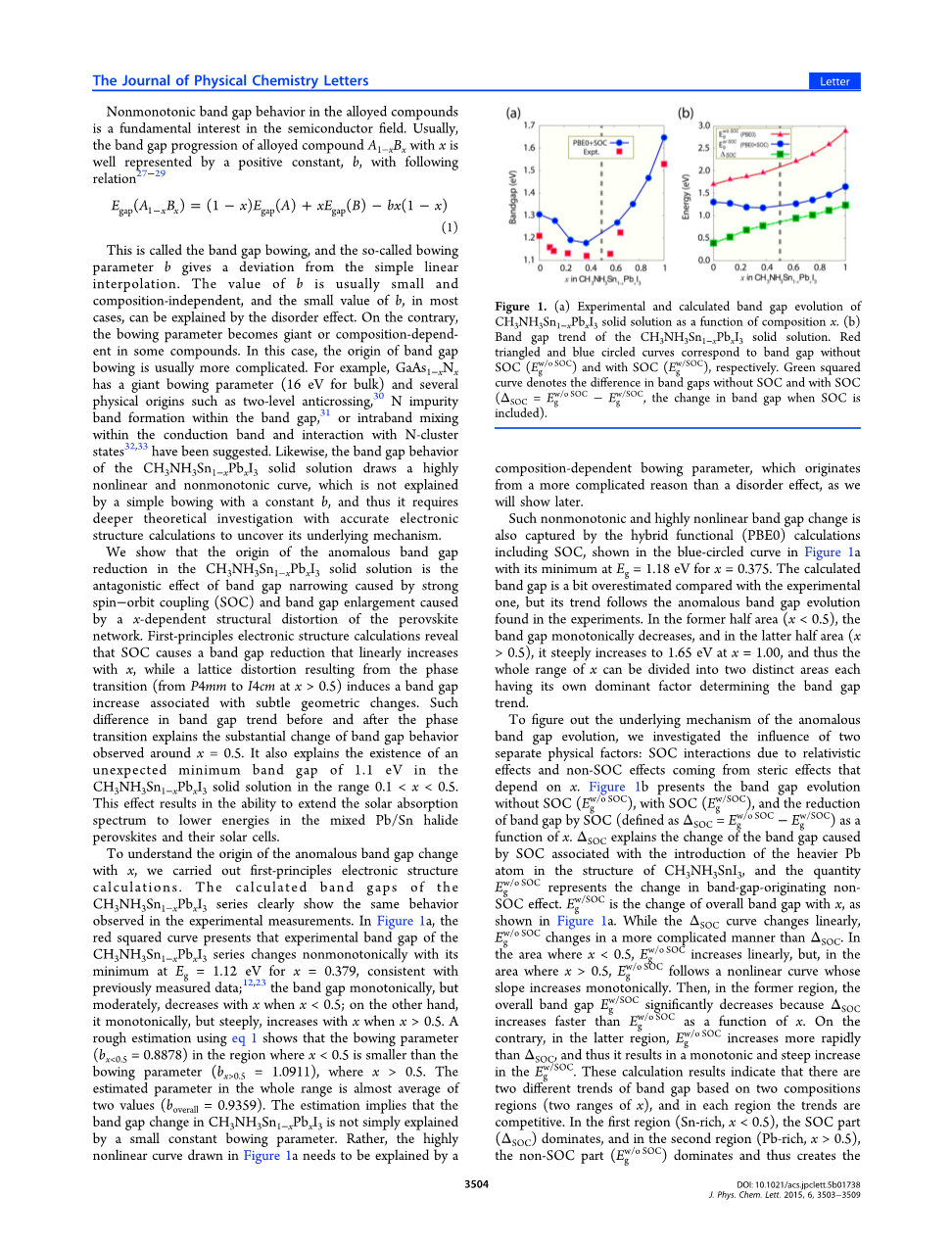
为了了解x的异常带隙变化的起源,我们实现了第一原理电子结构。通过CH3NH3Sn1-xPbxI3系列的分析,可以清楚地显示出在实验测量中观察到的相同的行为。在图1a中,红色平方曲线表明,CH3NH3Sn1-xPbxI3系列的实验带隙在x = 0.379处以Eg = 1.12eV的最小值非单调变化,与先前测量的数据一致; 带隙单调,但适度,当x lt;0.5时,x随x减小;另一方面,当xgt; 0.5时,它单调地,但陡峭地增加x。使用等式1的粗略估计表明弓形参数(bx lt;0.5 = 0.8878),其中x lt;0.5小于弯曲参数(bxgt; 0.5 = 1.0911),其中xgt; 0.5。整个范围内的估计参数几乎是两个值的平均值(boverall = 0.9359)。该估计意味着CH3NH3Sn1-xPbxI3中的带隙变化不是简单的解释通过小的恒定弯曲参数。相反,图1a中绘制的高度非线性曲线需要通过组合相关的弓形参数进行解释,该参数起源于比混乱效应更复杂的原因,我们稍后将会展示。
图1.(a)作为组成x的函数的CH3NH3Sn1-xPbxI3固溶体的实验和计算的带隙演变。(b)CH3NH3Sn1-xPbxI3固溶体的带隙趋势。红三角和蓝色圆圈曲线分别对应无SOC(Eg w/ oSOC)和SOC(Egw / SOC)的带隙。绿色平方曲线表示没有SOC和SOC的带隙(SOC = Eg w/ oSOC-Egw / SOC,包含SOC时带隙的变化)的差异。
这种非单调和高度非线性的带隙变化也由包括SOC的混合功能(PBE0)计算(图1a中蓝色圆圈曲线所示)捕获,对于x = 0.375,其最小值为Eg = 1.18eV。与实验相比,计算的带隙有点高估,但其趋势遵循实验中发现的异常带隙演化。在前半部分(x lt;0.5)中,带隙单调减小,而在后半部分(xgt; 0.5)中,x = 1.00时,陡峭增加到1.65 eV,因此x的整个范围可以分为分成两个不同的区域,每个区域都有自己的决定带隙趋势的主导因素。
为了找出异常带隙演化的潜在机制,我们研究了两种不同物理因素的影响:SOC相互作用由相对论效应和非SOC效应来自空间效应,取决于x。图1b表示出了SOC(Egw/ oSOC),SOC(Egw / SOC)和通过SOC(被定义为SOC = Eg w/ oSOC- Egw / SOC)的带隙的减小作为功能的带隙演化X。 SOC解释了带隙的变化通过与在CH3NH3SnI3的结构中引入较重的Pb原子相关的SOC,以及数量代表带隙发生非线性变化的变化,SOC效应Egw / SOC是整体带隙与x的变化,如图1a所示。当SOC曲线线性变化时,Egw/ oSOC以比SOC更加复杂的方式变化。 在x lt;0.5,Egw/ oSOC线性增加的区域,但是在xgt; 0.5的区域,Egw/ oSOC遵循斜率单调增加的非线性曲线。那么,在前一个地区,总体带隙Egw / SOC显著下降,因为SOC作为x的函数,比Egw/ oSOC增加更快。相反,在后一个地区,Egw/ oSOC比SOC增长更快,因此导致Egw / SOC的单调和急剧增加。这些计算结果表明有基于两个组成区域(x的两个范围)的两个不同的带隙趋势,并且在每个区域中,趋势是有竞争力的。在第一区域(富Sn,x lt;0.5),SOC部分(SOC)占主导地位,而在第二区域(富Pb,xgt; 0.5),非SOC部分(Egw/ oSOC)占主导地位,从而在CH3NH3Sn1-xPbxI3固溶体系列中产生跨越x的异常带隙演化。
带隙的非SOC部分的趋势与晶体对称性和结构几何的变化密切相关。实验上,我们发现CH3NH3Sn1-xPbxI3的晶体结构呈现x = 0.5附近的相变,从富Sn组成的特征的P4mm空间群(alpha;相)切换到I4cm空间群(beta;相)对于富含Pb的组合物来说是显性的。通过[MI6] 4-八面体沿垂直于晶体c轴的相反方向(异相倾斜)的旋转而引起空间群变化。旋转是立体声,其导致M-I-M角度的显着变化,其在CH3NH3PbI3(163.55°)中最显着,但随着Sn掺入Pb位点,效应逐渐减小(图中的红色平方曲线图2a)。
图2.(a)作为x的函数的M-I-M倾斜角的演变。由红色方块和蓝色圆圈定义的曲线分别对应于实验和计算结果。 (b,c)分别具有MI6八面体结构的x = 0.0(P4mm)和1.0(I4cm)的晶体结构。
为了比较,CH3NH3SnI3中的Sn-I-Sn角为177.4°。正如我们以前指出的,由笼状阳离子与钙钛矿笼尺寸的半径比决定的M-I-M角强烈地影响带隙.对于给定的金属(Pb,Sn等)M-I-M角度的线性偏差越大,能隙越大。这是因为线性M-I-M角度使得框架中的M-I-M原子之间的p轨道重叠最大化,给出最宽的带宽并因此产生最窄的带隙。该原理适用于所有AMX3卤化物钙钛矿。通过DFT优化的松弛晶体结构表现出与M-I-M倾斜角度相似的行为,实验确定倾斜角度。在图2a中,由蓝色圆圈曲线表示的计算数据表明,M-I-M角度在x = 1.0处具有与线性最大的偏差,随着Sn取代结构中的Pb而减小。然而,与实验结果相比,计算过高了alpha;相的倾斜角,而beta;相的角度很好。这种行为是由于在alpha;相中金属离子在八面体的中心移动的过高倾向。如图2b,c所示,八面体的畸变在alpha;相中比在beta;相中通过金属离子的中心更显着,因此alpha;相的计算倾斜角度的过高估计不表示[MI6] 4-八面体。该结果可归因于CH3相H 阳离子在alpha;相中的单向取向,这可以加强有机-无机杂化卤化物钙钛矿中的铁电极化.更详细的结构信息在支持信息中讨论。
在下一段中,我们进一步讨论了alpha;和beta;相的电子带结构的重要特征,以及它们如何影响中间组分的带隙变化。
如先前的研究所示,卤代钙钛矿的低能电子结构强烈地受到重金属如Sn和Pb的SOC的影响。因此,为了找出结构相变与异常带隙演化的关系,重要的是确定由Sn / Pb合金化引入的局部SOC强度的变化在多大程度上与alpha;相和beta;相的晶体对称性。作为alpha;和beta;相的代表,我们绘制了x = 0.0和1.0组成的电子带结构。图3a,b显示了左列中的状态(PDOS)的预测密度,中柱中没有SOC的电子带结构,以及x = 0.0和1.0的右侧列的SOC。 没有SOC时,CH3NH3SnI3的带隙由导带最小值(CBM)和由Sn-s和Ip轨道组成的一个杂化态由三个Sn p轨道状态(IpSnxgt;,IpS
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[141894],资料为PDF文档或Word文档,PDF文档可免费转换为Word
课题毕业论文、外文翻译、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。

